Simulation of rovibronic spectra for small molecules
Molecular spectroscopy experiments analyze the light that molecules absorb or emit. The collected data provide information on the species of the molecules present and about their abundances. Such information can be measured not only in the laboratory, but also in remote environments that are difficult to access such as the upper layers of the Earth's atmosphere, interstellar space, and the atmospheres of other planets, comets, and cool stars. Spectroscopic methods are thus ideally suited for remote sensing and play an important role in environmental research, in particular in the investigation of the Earth's atmosphere, and in astrophysics and -chemistry.
In order to assist the investigations of, for instance, interstellar space, the outer layers of cool stars, and the higher layers of the terrestrial atmosphere by remote sensing experiments (such as radio astronomy), we are carrying out simulations of the rovibronic spectra of small molecules that are present, or could be present, in space and/or in the atmosphere. For these simulations, we use methods that have been developed by our group (mostly in collaboration with other groups) during the last 25 years. In particular, we use the program systems MORBID (Morse Oscillator Rigid Bender Internal Dynamics), RENNER, DR (Double Renner), XY3, and TROVE (Theoretical ROtation-Vibration Energies). The simulations are based on ab initio potential-energy und dipole-moment surfaces that are calculated by our collaboration partners.

As an example we show here the rotational spectrum in the vibrational ground state of the HSOH molecule, simulated with the program TROVE at a temperature of 300 K.
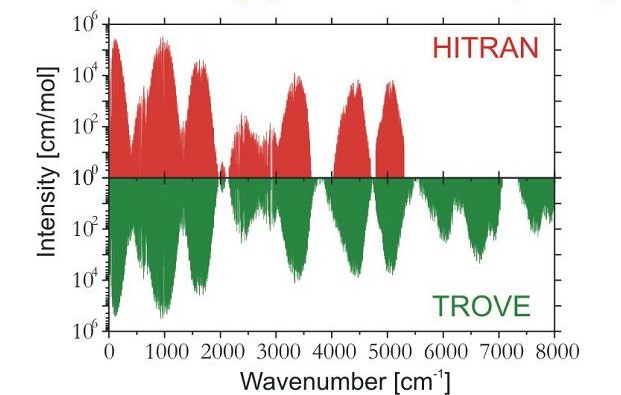
After we had developed (in collaboration with Dr. Sergei N. Yurchenko, Technical University Dresden, Germany, und Prof. Walter Thiel, Max Planck Institute for Coal Research, Mülheim/Ruhr, Germany) the program system TROVE (Theoretical ROtation-Vibration Energies) we became able to simulate the rotation-vibration spectrum of an arbitrary molecule in an isolated electronic state in variational calculations. We used TROVE to generate a so-called linelist (i.e., a catalogue of wavenumbers and intensities for as many transitions as possible) for ammonia NH3. The purpose of these very demanding calculations (made in collaboration Dr. Yurchenko, Prof. Thiel and Prof. Jonathan Tennyson, University College London, UK, on computers at University College London) is to assist the observation and analysis of comets and cool stars. We first generated a line line at the temperature T = 300 K. The diagram shows a comparison of this list with the entries of the spectroscopy database HITRAN. It is seen that the theoretical calculation reproduces the observed HITRAN lines very well and, in addition, it provides many weak lines that are missing in HITRAN since they have not been experimentally observed and assigned.
Towards the end of 2010, a line list for NH3 at T = 1500 K was generated; this list can be used for analysing the spectra of cool stars.
Publications on spectral simulations
B. Ostojić, P. R. Bunker, P. Schwerdtfeger, B. Assadollahzadeh, and Per Jensen: The predicted spectrum of the hypermetallic molecule MgOMg, Phys. Chem. Chem. Phys. 13, 7546–7553 (2011).
A. Yachmenev, S. N. Yurchenko, Per Jensen, O. Baum, T. F. Giesen, and W. Thiel: Theoretical rotation-torsion spectra of HSOH, Phys. Chem. Chem. Phys. 12, 8387 - 8397 (2010).
S. N. Yurchenko, M. Carvajal, A. Yachmenev, W. Thiel, and Per Jensen: A theoretical-spectroscopy, ab-initio-based study of the electronic ground state of 121SbH3, J. Q. S. R. T. 111, 2279-2290 (2010).
A. Yachmenev, S. Yurchenko, I. Paidarová, Per Jensen, W. Thiel, and S. Sauer: Thermal averaging of the indirect nuclear spin-spin coupling constants of ammonia: the importance of the large amplitude inversion mode, J. Chem. Phys. 132, 114305/1-15 (2010).
T. Hirano, V. Derpmann, U. Nagashima, and Per Jensen: Large amplitude bending motion in CsOH, studied through ab initio-based three-dimensional potential energy functions, J. Mol. Spectrosc. 263, 150-159 (2010)
B. Ostojić, Per Jensen, P. Schwerdtfeger, B. Assadollahzadeh, and P. R. Bunker: The predicted infrared spectrum of the hyperberyllium molecule BeOBe in its X1<//span>Σg+and a3Σu+ electronic states, J. Mol. Spectrosc. 263, 21-26 (2010)
T. Hirano, U. Nagashima, G. Winnewisser, and Per Jensen: Electronic structures and rovibronically averaged geometries of the X6A' and Ã6A'' states of FeOH, J. Chem. Phys. 132, 094303/1-10 (2010).
S. N. Yurchenko, R. J. Barber, A. Yachmenev, W. Thiel, Per Jensen, and J. Tennyson: A variationally computed T=300 K line list for NH3, J. Phys. Chem. A 113, 11845-11855 (2009).
S. N. Yurchenko, A. Yachmenev, W. Thiel, O. Baum, T. F. Giesen, V. V. Melnikov, and Per Jensen: An ab initio calculation of the vibrational energies and transition moments of HSOH, J. Mol. Spectrosc. 257, 57-65 (2009).
R. I. Ovsyannikov, Per Jensen, M. Yu. Tretyakov, and S. N. Yurchenko: On the Use of the Finite Difference Method in a Calculation of Vibration-Rotation Energies, Optika i Spektroskopiya 107, 236-243 (2009) [in Russian]. English translation as Optics and Spectroscopy 107, 221-227 (2009).
T. Hirano, P. R. Bunker, S. Patchkovskii, U. Nagashima, and Per Jensen: The Predicted Spectrum of FeOH in Its Renner-degenerate X6A' and Ã6A'' <//span>Electronic States, J. Mol. Spectrosc. 256, 45-52 (2009). Πg electronic state, J. Mol. Structure 795, 9-13 (2006).
V. V. Melnikov, Per Jensen, and T. Hirano: Calculation of Rovibronic Intensities for Triatomic Molecules in Double-Renner-degenerate Electronic States. Application to the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 130, 224105/1-9 (2009).
R. I. Ovsyannikov, W. Thiel, S. N. Yurchenko, M. Carvajal, and Per Jensen: PH3 revisited: Theoretical transition moments for the vibrational transitions below 7000 cm-1, J. Mol. Spectrosc. 252, 121-128 (2008).
R. I. Ovsyannikov, V. V. Melnikov, W. Thiel, Per Jensen, O. Baum. T. F. Giesen, and S. N. Yurchenko: Theoretical rotation-torsion energies of HSOH, J. Chem. Phys. 129, 154314/1-9 (2008).
R. I. Ovsyannikov, W. Thiel, S. N. Yurchenko, M. Carvajal, and Per Jensen: Vibrational energies of PH3 calculated variationally at the complete basis set limit, J. Chem. Phys. 129, 044309/1-8 (2008).
S. N. Yurchenko, W. Thiel, M. Carvajal, and Per Jensen: Ab initio potential energy surface, electric dipole moment, polarizability tensor, and theoretical rovibrational spectra in the electronic ground state of 14NH3+, Chem. Phys., 346, 146-159 (2008).
S. N. Yurchenko, W. Thiel, and Per Jensen: Theoretical ROVibrational Energies (TROVE): A robust numerical approach to the calculation of ro-vibrational energies for polyatomic molecules, J. Mol. Spectrosc. 245, 126-140 (2007).
P. R. Bunker, W. P. Kraemer, S. N. Yurchenko, W. Thiel, C. F. Neese, J. L. Gottfried, and Per Jensen: New potential energy surfaces for the X and A states of CH2+, Mol. Phys. 105, 1369-1376 (2007).
S. N. Yurchenko, M. Carvajal, W. Thiel, and Per Jensen: Ab initio dipole moment and theoretical rovibrational intensities in the electronic ground state of PH3, J. Mol. Spectrosc. 239, 71-87 (2006).
P. R. Bunker, R. Guérout, Z. J. Jakubek, Per Jensen, and S. N. Yurchenko: The rovibronic energies of the SiNSi radical in its X2
Z. J. Jakubek, P. R. Bunker, M. Zachwieja, S. G. Nakhate, B. Simard, S. N. Yurchenko, W. Thiel, and Per Jensen: A Dispersed Fluorescence and Ab Initio Investigation of the X2B1 and Ã2A1 Electronic States of the PH2 Molecule, J. Chem. Phys. 124, 094306/1-5 (2006).