The Renner Effect
The effects on the spectrum of electronic orbital and spin angular momentum in triatomic molecules is being investigated in collaboration with P. R. Bunker (Steacie Institute for Molecular Sciences, National Research Council of Canada, Ottawa), T. Hirano (Ochanomizu University, Tokyo, Japan), W. P. Kraemer (Max Planck Institute of Astrophysics, Garching, Germany) and others. This is generally termed the Renner effect. We initially developed a computer program RENNER with which we can calculate both the positions and intensities of the lines in a spectrum that arises from transitions between the two halves of a Renner state. Applications to free radicals and molecular ions are being undertaken using potential energy surfaces calculated by ab initio methods. During the last 15 years, we have predicted the electronic spectra of a very large number of triatomic molecules including the NH2+ and CH2+ ions, and these predictions have assisted in the search for the spectra of these molecules and their interpretation.
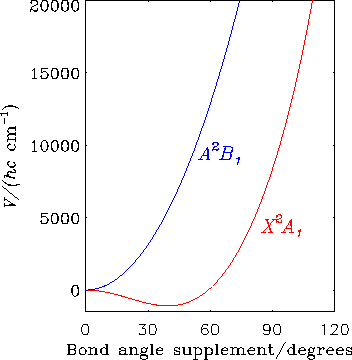
The diagram shows how for the CH2+ ion, the electronic states X2A1 and A2B1 are degenerate at linear geometries; the abscissa is the supplement of the H-C-H bond angle. These two electronic states are subject to the Renner effect.
Around Year 2000 it was conjectured for CH2+, on the basis of the interpretation of data obtained using the Coulomb explosion imaging (CEI) method, that there is a large nonadiabatic contribution to the low-lying wavefunctions beyond that coming from the Renner effect. We calculated the energies of the lowest excited electronic states and found, in agreement with results that were already in the literature, that the excited electronic states of CH2+ are at much too high an energy (greater than 6 eV) for such nonadiabatic interaction to be significant. To compare with the CEI results we calculate the Boltzmann averaged bending angle distribution using our previously calculated ab initio potential energy curves of the X/A pair of Renner interacting potentials, and make full allowance for the Renner effect in the calculation of the wavefunctions. This ab initio calculation leads to a distribution that is significant over a narrower range of bending angles than that originally obtained experimentally by the CEI method. A re-analysis of the experimental Coulomb-explosion data, however, revealed an inaccuracy in the interpretation of the experimental data. The re-interpreted data were in very good agreement with our simulation.
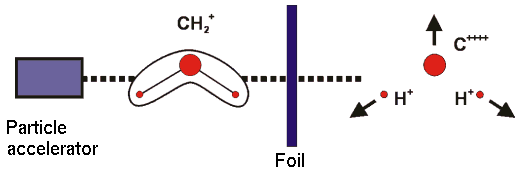
The diagram shows the principle of a CEI experiment. Molecular ions are accelerated and "shot" through a foil, where they lose several electrons. The remaining, highly unstable system "explodes" due to repulsive Coulomb forces, and by detection of the fragments the molecular geometry in the instant of the explosion can be determined.
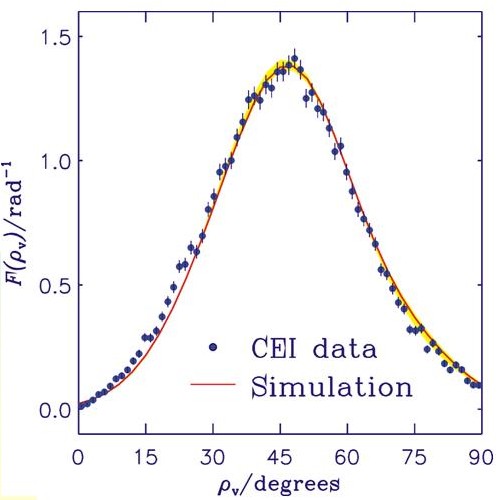
The figure shows the experimental CEI data points as dots and the result of our simulation as a solid curve. The angle ρv is the supplement of the C-H-C bond angle. The curve shown here is the result of applying so-called foil effects (i.e., broadening effects that result from the ions traversing the foil) to the "molecule-internal" curve obtained in our calculation. The foil effects are simulated by Monte Carlo methods. For experimental details, see L. Lammich, Diploma thesis, Faculty of Physics and Astronomy, Ruprecht-Karls-Universität, Heidelberg, 2001, and L. Lammich, H. Buhr, H. Kreckel, S. Krohn, M. Lange, D. Schwalm, R. Wester, A. Wolf, D. Strasser, D. Zajfman, Z. Vager, L. Abril, S. Heredia-Avalos, and R. Garcia-Molina, Phys. Rev. A 69, 062904 (2004). The Coulomb-explosion-imaging experiment was re-interpreted as a consequence of our theoretical results. This is a rare case of theory changing experiment.
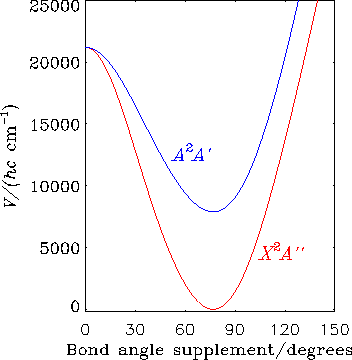
The HO2 molecule in the X2A'' and A2A' electronic states is the subject of further RENNER calculations. The diagram shows the bending potential energy curves in these two electronic states.
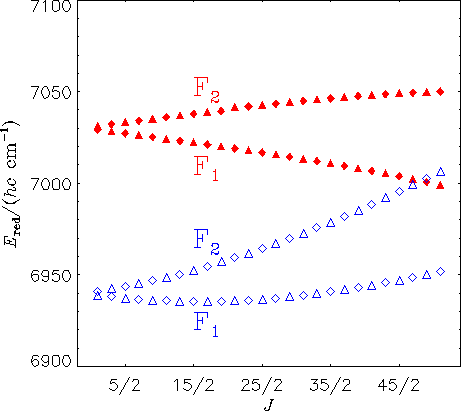
The diagram above shows reduced energies for the Ka = 0 states in the A(0,0,0) (filled triangles and diamonds) and X(1,1,2) (empty triangles and diamonds) vibronic states of HO2. A diamond represents a state with positive parity [symmetry A' in Cs(M)] and a triangle represents a state with negative parity [symmetry A'' in Cs(M)]. The calculations predict a local perturbation of the A(0,0,0) levels around J = 51/2, in good agreement with experimental findings of E. H. Fink and D. A. Ramsay [J. Mol. Spectrosc. 185, 304-324 (1997)]. Based on our theoretical calculations, the perturbing state can be identified as X(1,1,2).
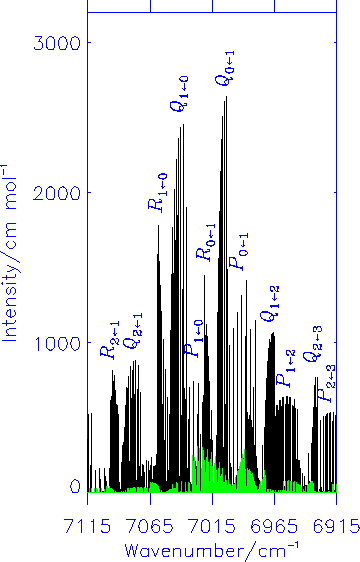
Fink and Ramsay [J. Mol. Spectrosc. 185, 304-324 (1997)] also found a number of lines with anomalous selection rules; apparently these transitions take place between states of the same parity indicating that they are magnetic dipole lines. Our theoretical calculations confirmed that in the X2A'' <- A2A' electronic transition of HOO, the magnetic dipole transitions have intensities typically around 1/10 of that of the electric dipole lines. The diagram shows the simulated electric dipole spectrum in black and the magnetic dipole spectrum in green.
The double Renner effect
In recent years, we have been concerned with the "double Renner effect" which we encounter when, because of isomerization, two linear geometries, ABC and BCA, say, become accessible to a triatomic molecule and the electronic energy is Renner-degenerate at both these geometries. In the framework of the Ph.D. study of Tina Erica Odaka, we developed a new computer program, DR, which calculated the rovibronic energies for a triatomic molecule in a double-Renner-degenerate electronic state. Vladlen V. Melnikov has later extended the DR program so that it can also simulate rovibronic spectra.
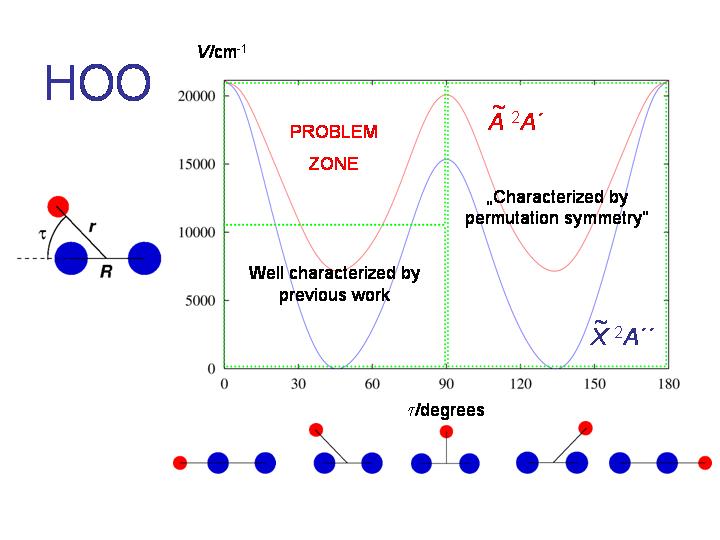
If we extend the RENNER-description of HO2 discussed above, this molecule affords an example of the double Renner effect. As seen in the diagram, the molecule is bent at equilibrium and has two equivalent equilibrium configurations separated by a barrier corresponding to a T-shaped geometry. Near each equilibrium geometry there is a linear configuration where the two lowest electronic states, X2A" and A2A', become energetically degenerate.
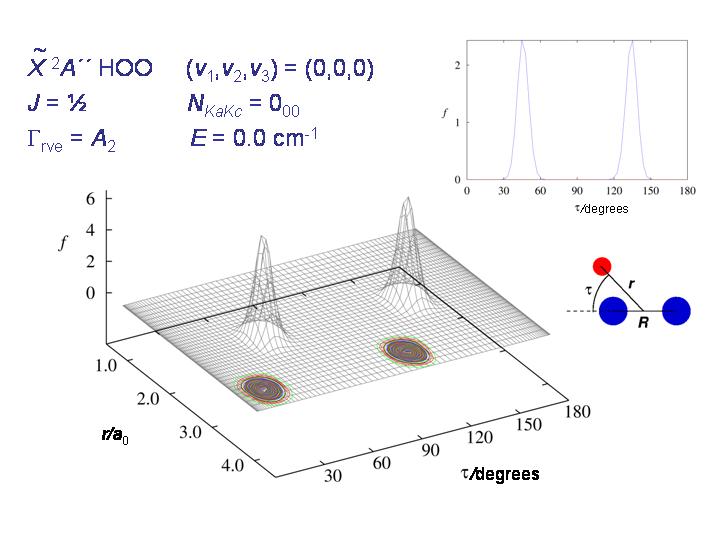
The diagram shows, for HO2, the probability distribution for the coordinates r and τ (see diagram) for the rovibronic ground state. The molecule is nicely localized to one of the two potential minima.
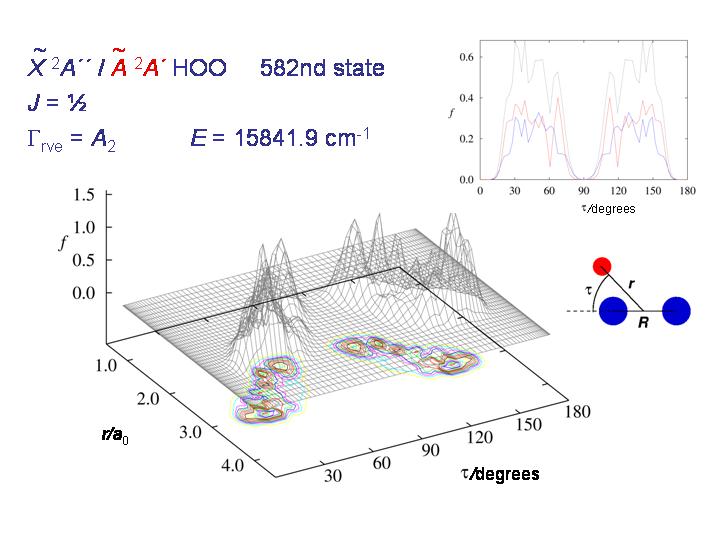
At higher energy, the molecule starts being able to tunnel through the barrier as seen in the diagram above.
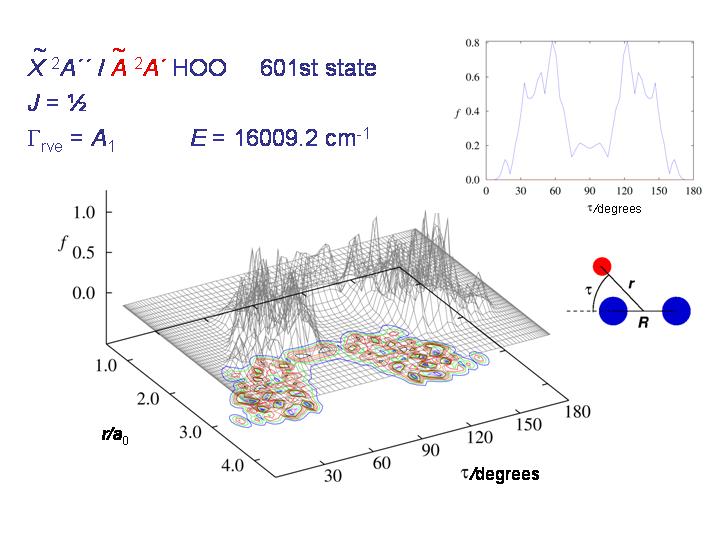
At still higher energy, the molecule can roam rather freely over the whole τ interval from 0 to 180 degrees and the distribution becomes "chaotic and beautiful", as T. E. Odaka put it in her 2004 Ph.D. thesis.
By means of the DR program we have also made extensive theoretical investigations of the first excited electronic state, A2Π, of the isomerizing double-Renner molecule MgNC/MgCN. For this molecule, we found theorerical evidence of so-called Renner-driven isomerization, where the transfer from the MgNC minimum to the MgCN minimum on the A2Π potential energy surfaces is accompanied by an "electronic transition" between the two components of the Π state.
Publications about the Renner Effect
(215) T. Hirano, U. Nagashima, and Per Jensen: Computational Molecular Spectroscopy of X2Π NCS: Electronic properties and ro-vibrationally averaged structure, J. Mol. Spectrosc. 346, 4–12 (2018). DOI: 10.1016/j.jms.2017.12.011
(214) J. Freund, S. C. Galleguillos Kempf, Per Jensen, U. Nagashima, and T. Hirano: Computational Spectroscopy of NCS in the Renner-degenerate electronic state X2Π, J. Mol. Spectrosc. 345, 31–38 (2018). DOI: 10.1016/j.jms.2017.11.010
(172) T. Hirano, U. Nagashima, G. Winnewisser, and Per Jensen: Electronic structures and rovibronically averaged geometries of the X6A' and Ã6A'' states of FeOH, J. Chem. Phys. 132, 094303/1-10 (2010).
(168) T. Hirano, P. R. Bunker, S. Patchkovskii, U. Nagashima, and Per Jensen: The Predicted Spectrum of FeOH in Its Renner-degenerate X6A' and Ã6A'' Electronic States, J. Mol. Spectrosc. 256, 45-52 (2009).
(166) V. V. Melnikov, Per Jensen, and T. Hirano: Calculation of Rovibronic Intensities for Triatomic Molecules in Double-Renner-degenerate Electronic States. Application to the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 130, 224105/1-9 (2009).
(161) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: Computational Molecular Spectroscopy for X2Δ NiCN: Large Amplitude Bending Motion, J. Mol. Spectrosc. 250, 33-43 (2008).
(159) T. Hirano, R. Okuda, U. Nagashima, K. Tanaka, and Per Jensen: Ab Initio Molecular Orbital Study of Ground and Low-Lying Electronic States of NiCN, Chem. Phys., 346, 13-22 (2008).
(158) V. V. Melnikov, T. E. Odaka, Per Jensen, and T. Hirano: The Double Renner Effect in the X2A'' and Ã2A' Electronic States of HO2, J. Chem. Phys. 128, 114316/1-10 (2008).
(156) P. R. Bunker, W. P. Kraemer, S. N. Yurchenko, W. Thiel, C. F. Neese, J. L. Gottfried, and Per Jensen: New potential energy surfaces for the X and A states of CH2+, Mol. Phys. 105, 1369-1376 (2007).
(154) T. Hirano, M. Amano, Y. Mitsui, S. S. Itono, R. Okuda, U. Nagashima, and Per Jensen: A Theoretical Study of FeCN in the 6Δ Electronic Ground State, J. Mol. Spectrosc. 243, 267-279 (2007).
(153) T. Hirano, R. Okuda, U. Nagashima, Y. Nakashima, K. Tanaka, and Per Jensen: A theoretical study of BrCN+ in the 2Π electronic ground state: Large amplitude bending motion, J. Mol. Spectrosc. 243, 202-218 (2007).
(152) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: Ab Initio Molecular Orbital Study of Ground and Low-Lying Electronic States of CoCN, J. Chem. Phys. 127, 014303/1-7 (2007).
(151) T. Hirano, R. Okuda, U. Nagashima, and Per Jensen: A Theoretical Study of CoCN in the 3Φ Electronic Ground State, Mol. Phys. 105, 599-611 (2007).
(150) T. E. Odaka, V. V. Melnikov, Per Jensen, T. Hirano, B. Lang, and P. Langer: Theoretical Study of the Double Renner Effect for Ã2Π MgNC/MgCN: Higher Excited Rovibrational States, J. Chem. Phys. 126, 094301/1-9 (2007).
(146) T. E. Odaka, Per Jensen, and T. Hirano: The Double Renner Effect: A Theoretical Study of the MgNC/MgCN Isomerization in the Ã2Π Electronic State, J. Mol. Structure 795, 14-41 (2006).
(145) P. R. Bunker, R. Guérout, Z. J. Jakubek, Per Jensen, and S. N. Yurchenko: The rovibronic energies of the SiNSi radical in its X2Πg electronic state, J. Mol. Structure 795, 9-13 (2006).
(144) T. Hirano, R. Okuda, U. Nagashima, V. Špirko, and Per Jensen: A Theoretical Study of FeNC in the 6Δ Electronic Ground State, J. Mol. Spectrosc. 236, 234-247 (2006).
(128) S. Wu, Y. Chen, X. Yang, Y. Guo, Y. Liu, Y. Li, R. J. Buenker, and Per Jensen: Vibronic Transition Moments and Line Intensities in H2O+, J. Mol. Spectrosc. 225, 96-106 (2004).
(126) Per Jensen, W. P. Kraemer, and P. R. Bunker, Transition moments and NH2 cometary spectra, Mol. Phys. 101, 613-622 (2003).
(124) P. R. Bunker, W. P. Kraemer, Per Jensen, Y.-C. Lee, and Y.-P. Lee, The Matrix Isolation Spectrum of the CH2+ Ion, J. Mol. Spectrosc. 216, 419-423 (2002).
(123) T. E. Odaka, T. Hirano, and Per Jensen: A Theoretical Study of Ã2Π MgCN, J. Mol. Spectrosc. 216, 379-396 (2002).
(122) T. Hirano, K. Ishii, T. E. Odaka, and Per Jensen: A Theoretical Study of MgNC and MgCN in the X2Σ+ Electronic State, J. Mol. Spectrosc. 215, 42-57 (2002).
(120) T. E. Odaka, T. Hirano, and Per Jensen: An Ab Initio Study of the Ã2Π and the Ã2Π ← X2Σ+ Electronic Transition of MgNC, J. Mol. Spectrosc. 211, 147-161 (2002).
(119) Per Jensen, T. E. Odaka, W. P. Kraemer, T. Hirano, and P. R. Bunker: The Renner Effect in Triatomic Molecules with Application to CH2+, MgNC and NH2, Spectrochimica Acta Part A 58, 763-794 (2002).
(117) S. N. Yurchenko, Per Jensen, Y. Li, R. J. Buenker, and P. R. Bunker: The Near Ultraviolet Band System of Singlet Methylene, J. Mol. Spectrosc. 208, 136-143 (2001).
(116) P. R. Bunker, M.-C. Chan, W. P. Kraemer, and Per Jensen: Predicted Rovibronic Spectra of CH2+ and CD2+, Chem. Phys. Lett. 341, 358-362 (2001).
(115) Per Jensen, R. J. Buenker, J.-P, Gu, G. Osmann and P. R. Bunker: Refined Potential Energy Surfaces for the X2A'' and A2A' Electronic States of the HO2 Molecule, Can. J. Phys. 79, 641-652 (2001).
(111) G. Osmann, P. R. Bunker, W. P. Kraemer, and Per Jensen: Coulomb Explosion Imaging: The CH2+, H2O+ and NH2+ Ions as Benchmarks, Chem. Phys. Lett. 318, 597-606 (2000).
(108) G. Osmann, P. R. Bunker, W. P. Kraemer, and Per Jensen: Coulomb Explosion Imaging and the CH2+ Molecule, Chem. Phys. Lett., 309, 299-306 (1999).
(105) G. Osmann, P. R. Bunker, Per Jensen, R. J. Buenker, J.-P. Gu, and G. Hirsch: A Theoretical Investigation of the Renner Interactions and Magnetic Dipole Transitions in the A - X Electronic Band System of HO2, J. Mol. Spectrosc., 197, 262-274 (1999).
(103) J.-P. Gu, G. Hirsch, R. J. Buenker, M. Brumm, G. Osmann, P. R. Bunker and P. Jensen: A theoretical study of the absorption spectrum of singlet CH2, J. Mol. Struct., 517, 247-264 (2000).
(101) G. Osmann, P. R. Bunker, P. Jensen and W. P. Kraemer: An Ab Initio Study of the NH2+ Absorption Spectrum. J. Mol. Spectrosc. 186, 319 (1997)
(100) G. Osmann, P. R. Bunker, P. Jensen and W. P. Kraemer: A Theoretical Calculation of the Absorption Spectrum of CH2+. Chem. Phys. 225, 33 (1997).
(85) J.-P. Gu, R. J. Buenker, G. Hirsch, P. Jensen and P. R. Bunker: An ab initio calculation of the BH2- rovibronic energies: a very small singlet-triplet splitting. J. Mol. Spectrosc. 178, 172 (1996).
(79) M. Kolbuszewski, P. R. Bunker, W. P. Kraemer, G. Osmann and P. Jensen: An ab initio calculation of the rovibronic energies of the BH2 molecule. Mol. Phys. 88, 105 (1996).